3 Questions: On biology and medicine’s “data revolution”
Caroline Uhler is an Andrew (1956) and Erna Viterbi Professor of Engineering at MIT; a professor of electrical engineering and computer science in the Institute for Data, Science, and Society (IDSS); and director of the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard, where she is also a core institute and scientific leadership team member.
Uhler is interested in all the methods by which scientists can uncover causality in biological systems, ranging from causal discovery on observed variables to causal feature learning and representation learning. In this interview, she discusses machine learning in biology, areas that are ripe for problem-solving, and cutting-edge research coming out of the Schmidt Center.
Q: The Eric and Wendy Schmidt Center has four distinct areas of focus structured around four natural levels of biological organization: proteins, cells, tissues, and organisms. What, within the current landscape of machine learning, makes now the right time to work on these specific problem classes?
A: Biology and medicine are currently undergoing a “data revolution.” The availability of large-scale, diverse datasets — ranging from genomics and multi-omics to high-resolution imaging and electronic health records — makes this an opportune time. Inexpensive and accurate DNA sequencing is a reality, advanced molecular imaging has become routine, and single cell genomics is allowing the profiling of millions of cells. These innovations — and the massive datasets they produce — have brought us to the threshold of a new era in biology, one where we will be able to move beyond characterizing the units of life (such as all proteins, genes, and cell types) to understanding the `programs of life’, such as the logic of gene circuits and cell-cell communication that underlies tissue patterning and the molecular mechanisms that underlie the genotype-phenotype map.
At the same time, in the past decade, machine learning has seen remarkable progress with models like BERT, GPT-3, and ChatGPT demonstrating advanced capabilities in text understanding and generation, while vision transformers and multimodal models like CLIP have achieved human-level performance in image-related tasks. These breakthroughs provide powerful architectural blueprints and training strategies that can be adapted to biological data. For instance, transformers can model genomic sequences similar to language, and vision models can analyze medical and microscopy images.
Importantly, biology is poised to be not just a beneficiary of machine learning, but also a significant source of inspiration for new ML research. Much like agriculture and breeding spurred modern statistics, biology has the potential to inspire new and perhaps even more profound avenues of ML research. Unlike fields such as recommender systems and internet advertising, where there are no natural laws to discover and predictive accuracy is the ultimate measure of value, in biology, phenomena are physically interpretable, and causal mechanisms are the ultimate goal. Additionally, biology boasts genetic and chemical tools that enable perturbational screens on an unparalleled scale compared to other fields. These combined features make biology uniquely suited to both benefit greatly from ML and serve as a profound wellspring of inspiration for it.
Q: Taking a somewhat different tack, what problems in biology are still really resistant to our current tool set? Are there areas, perhaps specific challenges in disease or in wellness, which you feel are ripe for problem-solving?
A: Machine learning has demonstrated remarkable success in predictive tasks across domains such as image classification, natural language processing, and clinical risk modeling. However, in the biological sciences, predictive accuracy is often insufficient. The fundamental questions in these fields are inherently causal: How does a perturbation to a specific gene or pathway affect downstream cellular processes? What is the mechanism by which an intervention leads to a phenotypic change? Traditional machine learning models, which are primarily optimized for capturing statistical associations in observational data, often fail to answer such interventional queries.There is a strong need for biology and medicine to also inspire new foundational developments in machine learning.
The field is now equipped with high-throughput perturbation technologies — such as pooled CRISPR screens, single-cell transcriptomics, and spatial profiling — that generate rich datasets under systematic interventions. These data modalities naturally call for the development of models that go beyond pattern recognition to support causal inference, active experimental design, and representation learning in settings with complex, structured latent variables. From a mathematical perspective, this requires tackling core questions of identifiability, sample efficiency, and the integration of combinatorial, geometric, and probabilistic tools. I believe that addressing these challenges will not only unlock new insights into the mechanisms of cellular systems, but also push the theoretical boundaries of machine learning.
With respect to foundation models, a consensus in the field is that we are still far from creating a holistic foundation model for biology across scales, similar to what ChatGPT represents in the language domain — a sort of digital organism capable of simulating all biological phenomena. While new foundation models emerge almost weekly, these models have thus far been specialized for a specific scale and question, and focus on one or a few modalities.
Significant progress has been made in predicting protein structures from their sequences. This success has highlighted the importance of iterative machine learning challenges, such as CASP (critical assessment of structure prediction), which have been instrumental in benchmarking state-of-the-art algorithms for protein structure prediction and driving their improvement.
The Schmidt Center is organizing challenges to increase awareness in the ML field and make progress in the development of methods to solve causal prediction problems that are so critical for the biomedical sciences. With the increasing availability of single-gene perturbation data at the single-cell level, I believe predicting the effect of single or combinatorial perturbations, and which perturbations could drive a desired phenotype, are solvable problems. With our Cell Perturbation Prediction Challenge (CPPC), we aim to provide the means to objectively test and benchmark algorithms for predicting the effect of new perturbations.
Another area where the field has made remarkable strides is disease diagnostic and patient triage. Machine learning algorithms can integrate different sources of patient information (data modalities), generate missing modalities, identify patterns that may be difficult for us to detect, and help stratify patients based on their disease risk. While we must remain cautious about potential biases in model predictions, the danger of models learning shortcuts instead of true correlations, and the risk of automation bias in clinical decision-making, I believe this is an area where machine learning is already having a significant impact.
Q: Let’s talk about some of the headlines coming out of the Schmidt Center recently. What current research do you think people should be particularly excited about, and why?
A: In collaboration with Dr. Fei Chen at the Broad Institute, we have recently developed a method for the prediction of unseen proteins’ subcellular location, called PUPS. Many existing methods can only make predictions based on the specific protein and cell data on which they were trained. PUPS, however, combines a protein language model with an image in-painting model to utilize both protein sequences and cellular images. We demonstrate that the protein sequence input enables generalization to unseen proteins, and the cellular image input captures single-cell variability, enabling cell-type-specific predictions. The model learns how relevant each amino acid residue is for the predicted sub-cellular localization, and it can predict changes in localization due to mutations in the protein sequences. Since proteins’ function is strictly related to their subcellular localization, our predictions could provide insights into potential mechanisms of disease. In the future, we aim to extend this method to predict the localization of multiple proteins in a cell and possibly understand protein-protein interactions.
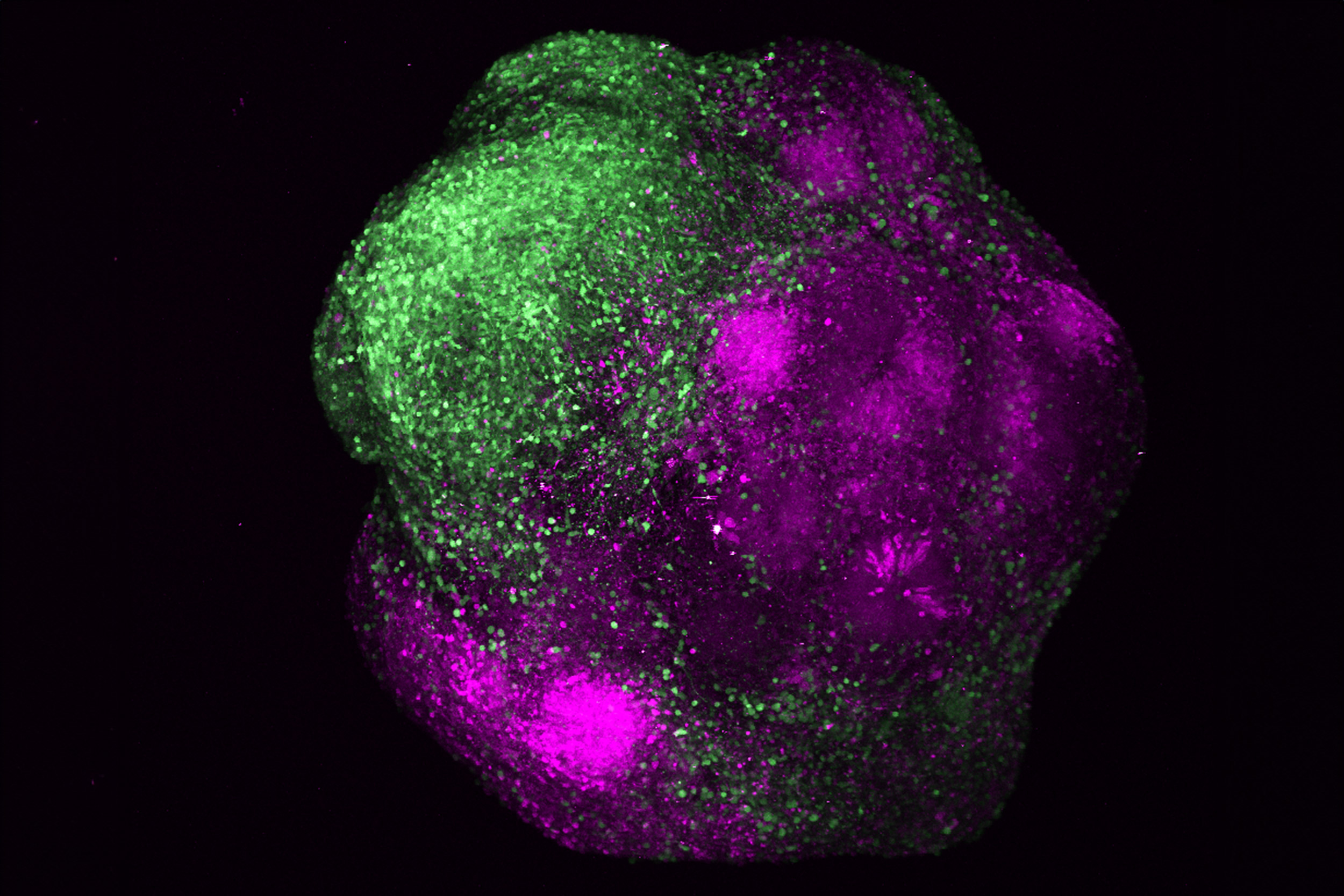
Together with Professor G.V. Shivashankar, a long-time collaborator at ETH Zürich, we have previously shown how simple images of cells stained with fluorescent DNA-intercalating dyes to label the chromatin can yield a lot of information about the state and fate of a cell in health and disease, when combined with machine learning algorithms. Recently, we have furthered this observation and proved the deep link between chromatin organization and gene regulation by developing Image2Reg, a method that enables the prediction of unseen genetically or chemically perturbed genes from chromatin images. Image2Reg utilizes convolutional neural networks to learn an informative representation of the chromatin images of perturbed cells. It also employs a graph convolutional network to create a gene embedding that captures the regulatory effects of genes based on protein-protein interaction data, integrated with cell-type-specific transcriptomic data. Finally, it learns a map between the resulting physical and biochemical representation of cells, allowing us to predict the perturbed gene modules based on chromatin images.
Furthermore, we recently finalized the development of a method for predicting the outcomes of unseen combinatorial gene perturbations and identifying the types of interactions occurring between the perturbed genes. MORPH can guide the design of the most informative perturbations for lab-in-a-loop experiments. Furthermore, the attention-based framework provably enables our method to identify causal relations among the genes, providing insights into the underlying gene regulatory programs. Finally, thanks to its modular structure, we can apply MORPH to perturbation data measured in various modalities, including not only transcriptomics, but also imaging. We are very excited about the potential of this method to enable the efficient exploration of the perturbation space to advance our understanding of cellular programs by bridging causal theory to important applications, with implications for both basic research and therapeutic applications.

© Photo: Jiin Kang















")
















































































 and lips (blue) before and after amputation")

















































































")
")








































")














